![[IUCr Home Page]](iucrhom2.gif)
![[Commission Home Page]](cpd.gif)
|
|
The IUCr-CPD Homepage is at http://www.iucr.org/iucr-top/comm/cpd/
BGMN - a New Fundamental Parameters Based Rietveld Program for Laboratory X-ray Sources, it's Use in Quantitative Analysis and Structure InvestigationsJ. Bergmann(*), P. Friedel(+), R. Kleeberg(=)(*) Ludwig-Renn-Allee 14, 01217 Dresden, Germany E-mail: bergmann@rcs.urz.tu-dresden.de (+)Institute of Polymer Research Dresden e.V., Hohe Str. 6, 01069 Dresden, Germany. E-mail: friedel@orion.ipfdd.de (=)University of Mining and Technology, Mineralogical Institute,Brennhausgasse 14, D-09595 Freiberg, Germany. E-mail: kleeberg@mineral.tu-freiberg.de http://www.mineral.tu-freiberg.de/mineralogie/bgmn/index.html
Common Problems of Rietveld ProgramsRietveld analyses can be executed by a lot of programs. Many functions were added since the first publication.A special problem for laboratory X-ray sources is the profile model [8]. The foregoing developers paid special attention to extend the profile model enabling the user to describe the peak shape within a wide angular range as exact as possible. In spite of all efforts, it was not possible to introduce a universal, precise profile model easy to be used until now. For that reason, the following measuring rule had to be fulfilled: Use narrow axial divergence collimators (Soller-collimator) to adapt the peak shape by simple analytical functions over the entire angular range. Even then difficulties often appeared in the case of peaks with 2 * angles less than 15 degrees. The so- called u-v-w parameters [7] are widely used for profile shape description. These three (or more) parameters must be fitted in conjunction with the crystallographic model parameters. Adaptation results in parameter correlation. It is a main source of divergence of the optimization algorithm, incorrect minima and program crashes. In addition, the wide-spread Rietveld programs need a lot of intuition for operation: Having declared an unfavorable set of parameters, the Rietveld programs react very sensitively. As a rule, they breakdown with an error inside the numerical library. In this case, the calculation which had been terminated compulsorily must be restarted from the beginning. This termination results from the use of simple optimization algorithms which cannot consider the physically reasonable ranges of parameters.
BGMN's Solutions to these ProblemsNew Profile Model
See: http://www.mineral.tu-freiberg.de/mineralogie/bgmn/index.html New Refinement Algorithm for Reliable Convergence The nonlinear least square algorithm is designed basing on a practical book by [23]. Within the central, linear part of the algorithm, a simple solution of the equation system above the Hesse matrix is replaced by the RG-CD algorithm (Restricted Gradient-Conjugated Direction) described in [22]. In its complete version, the algorithm supplies any in-equation constraints for linear combinations of parameters. Lower and upper parameter limits can be optionally defined by means of the simplified version used in BGMN [24].
New Features of BGMNDescription and Correction of Preferred Orientation To overcome the well-known problems caused by the March function [9], Järvinen has used spherical harmonics firstly [16]. We use a modified version of spherical harmonics until 10th order which can also be applied for samples of sophisticated orientation distributions. Parameter correlation and incorrect results can be avoided by defining the suitable order of the spherical harmonics resp. by the automatic reducing of the order depending on the measured intensity. Real Structure Functions The influence of different real structures on the scattering functions of polycrystals has been studied in detail by [18]. In the case of dislocations, he made the conclusion of gaussian peaks. Its width (squared variance) should be proportional to dislocation density as well as to length of scattering vector. Basing on [5], [20] has checked BGMN's model for valid crystallite size distribution. Equivalent to [5], he found the following formula:
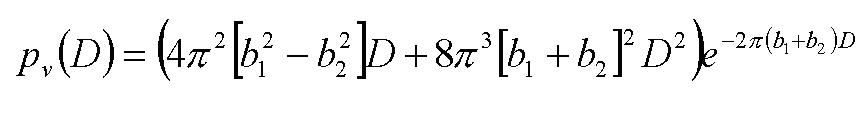 (1) (1) withpv = volume percentage of columns having a length D; in parallel to scattering vector. b1 = width parameter (half FWHM) of the simple Lorentzian part of the size broadening. b2 = width (standard deviation *) of the quadratic Lorentzian part of the size broadening. According to this we assume a Lorentz width b1 independent from the length of the scattering vector and a squared Lorentzian width b2 * b1. Both influences are greatly discussed in real structure literature, so the default formulae for peak widths considers both. Of course, one may assume other formulae. Molecular crystals (rigid bodies) New functions describing that lattice positions are placed by molecules instead of atoms were introduced. In cases of molecules, rotational parameters appear in addition to the translational ones. Based on the extended functions for molecules, we can define modifications of valence angles, torsion around bondings as well as stretching/compressing within molecules. Free programmability If the functions integrated are not sufficient to solve the special problem, define new parameters and dependencies by means of the formula interpreter inside the program. This way, it is easy to describe parameter couplings which are not part of the standard program capacity (e.g. between different atomic positions). Error analysis Beside of the well-known R values, BGMN calculates random error for all parameters and arbitrary functions of them. Therefore, one can declare arbitrary functions as so-called GOAL's. For every GOAL, value and ESD are calculated. Quantitative Phase AnalysisBecause of the excellent stability of the algorithm, BGMN enables routine quantitative phase analysis without the time-consuming work of programming "refinement strategies" or "analytical tasks" for new sample groups. Some special methods to use the BGMN features in QPA are:
Example of Routine Phase AnalysisAn example of routine analysis will be presented here. The sample is a commercial slate, used in reconstruction of historical buildings. It was prepared by stepwise grinding and sieving smaller 20 µm and packing into standard front-loading sample holder. Special problems are the strong PO and the Mg-Fe- substitution in chlorite minerals. The refinement starts without any background separation. The cell parameters of all phases present had to be refined. Substitution of Mg-Fe at 3 positions in chlorite, Ca- Fe and Mg-Fe substitution in ankerite and the K+ occupation factor in muscovite were also refined. Anisotropic line broadening models for muscovite and chlorite were used. Isotropic crystallite size broadening model was introduced for all other phases, for ankerite additional microstrain broadening was assumed. The program had to refine 75 parameters at all. A PentiumII 200MHz processor needed 5 minutes 6 seconds to fit 2467 measuring values and 915 peaks without any user interaction. The complex PO starting models of albite, microcline and ankerite have been reduced automatic to isotropy. The iron substitution in ankerite was calculated to the predefined limits. The quantitative results are given in table 1.
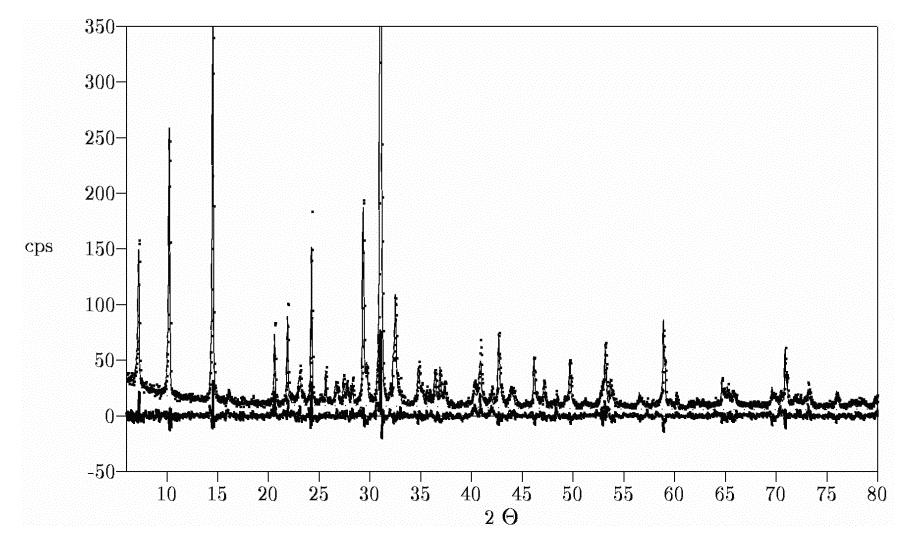 Figure 1: Rietveld refinement plot of slate sample. Co-K* radiation, Rwp=13.81%, Rexp=9.85%, Durbin- Watson d=1.22 Table 1: Results of slate quantitative analysis
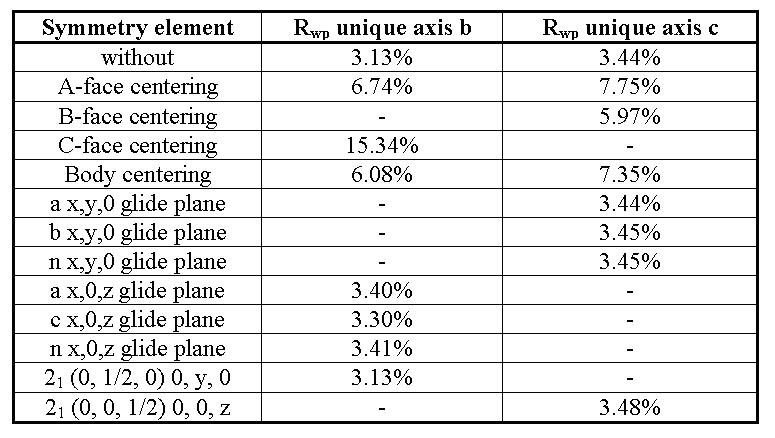
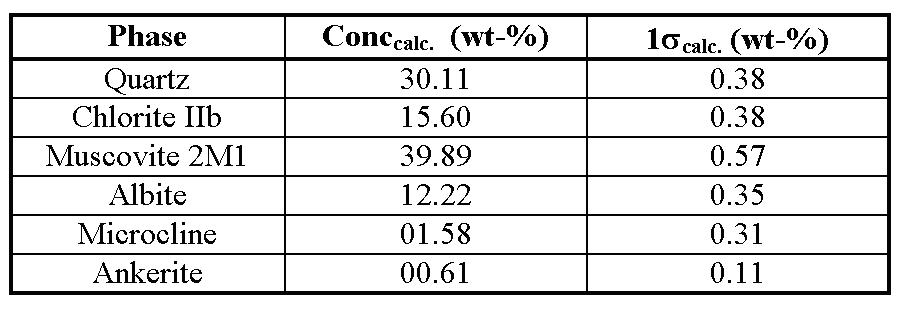 Note that the ankerite concentration calculated is near the detection limit but may be significant. The calculated individual PO correction factors of 00l chlorite and muscovite reflections are about 2.5. This is common value for layer silicates in front loading technique. Structure Refinement of Organic SolidsUsing the integrated formula interpreter, solid state structure refinement of organics is very easy to do by a four step procedure. Especially for polymers structure proposals are performed with reliability factors below 10% ([9],[10],[11],[12]). Here we present an overview about (IPA-HQ)n results [9]. Structureless Approximation The first step, the structureless approximation, results in unit cell parameters and information about symmetry operators. Table 2: Test of symmetry operators for (IPA-HQ)n
 Molecular Chain Model A molecule can be described by means of a graph tree model ([6]) using internal coordinates, recalculate the cartesian ones, shift it to the origin and orient it to one of the cartesian planes with one of its mean axis of inertia. Defining the parameters for global movement (rho x,y,z and SP vector) and internal conformation (bond length, bond angle and torsion angle) and the bondings between the atoms, the refinement procedure can be started.
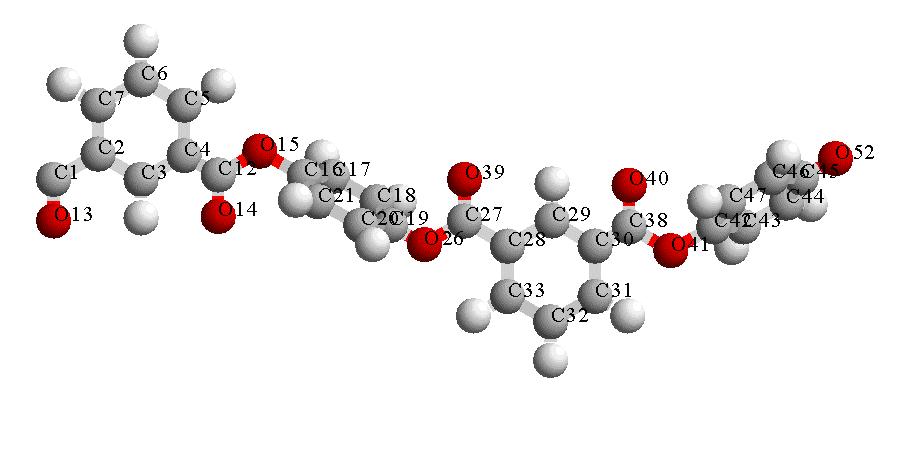 Figure 2: Asymmetric unit of (IPA-HQ)n (gray = C, light gray = H, dark = O) Empirical Force Field Model The combination of XPD Rietveld refinement with empirical force field energy minimization can favor a given approximation result [11], including nonbonding and penalty functions. Nonbonding terms like Lennard Jones potential avoid overlapping of atoms. Penalties enable a correct chain continuation [9], [10] or ring closures [11],[12]. Table 3: Constants of atom types for nonbonding interactions used for (IPA-HQ)n
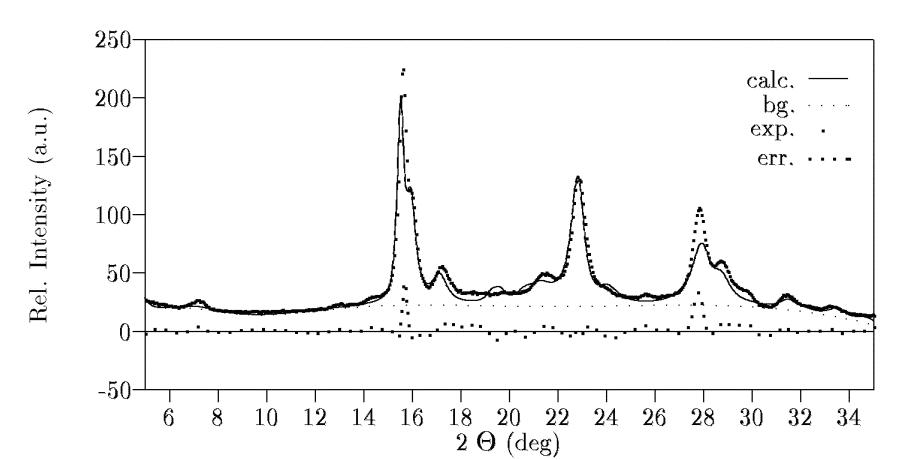 Table 4: Constants of penalty functions for bonding interactions used for (IPA-HQ)n
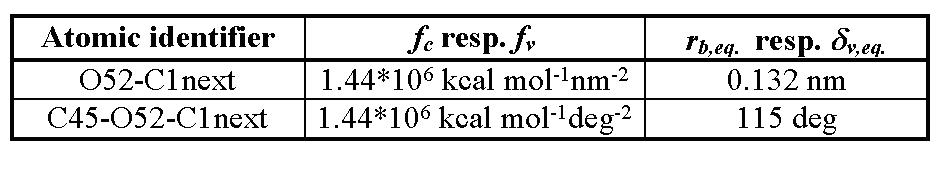 Other BGMN features used Major effects for improvement to the reliability factor can be achieved, since sample dependent effects would be respected. These are at first a preferred orientation of the crystallites, at second respecting possible micro strains in anisotropic form or at third special Debye-Waller factors for special atom types, which can be anisotropic also. The effects are decreasing in the called series. But note: Applying of these features should only be done, if one can be sure having the best result of approximation without. The summary of results can be seen in table 5. We also added the obtained scattering pattern including the approximation in figure 3. All results (the fractional coordinates and the structure factors F ) are published in a previous paper [17]. An optical expression can be seen at [13]. But note, this can be a structure proposal only because of the available experimental information. Table 5: Summary of results of structure investigations of (IPA-HQ)n

Figure 3: Approximation of (IPA-HQ)n internal structure parameter SummaryBGMN is a newly developed Rietveld program with many advantageous features like
This unique feature list enables BGMN to solve different problems. Until now, BGMN was used successfully in quantitative analysis and structure investigations of organic solids. PostscriptumThe authors wish to thank L.M.D. Cranswick for inviting to publish this paper in CPD Newsletters. They ask to apologize for it's formality and length: It has to been written during a single week, only.References
|

{kind=link}
{kind=link}