![[IUCr Home Page]](iucrhom2.gif)
![[Commission Home Page]](cpd.gif)
|
|
The IUCr-CPD Homepage is at http://www.iucr.org/iucr-top/comm/cpd/
Solving Crystal Structures Ab Initio Using Powder Diffraction DataJames A. KadukAmoco Corporation, P.O. Box 3011 MC F-9, Naperville IL 60566 USA. E-mail: kaduk@amoco.com; WWW: http://www.amoco.com Many technologically-important materials do not form the single crystals "necessary’ for determining their solid state structures using conventional crystallographic techniques. Knowledge of the crystal structure of a material makes it possible to rationalize/explain/predict many other properties, and facilitates the use of diffraction techniques to obtain morphological and mesostructural information from the powder data. Having the structure "in hand" when it is needed saves time and money; the effort of determining crystal structures thus is a sort of "intellectual capital investment", and can be justified as such. Solution of a crystal structure "asks a lot" from the powder data. The powder crystallographer is wise to use all information available about the compound. Such information includes chemical connectivity, local coordination, and stoichiometry. For small organic molecules we generally have the advantage that the molecular structure is known from spectroscopic techniques; it is "only" the crystal structure which must be determined. Several different techniques for solving structures using powder data are described here.
Dimethyl 2,6-naphthalenedicarboxylate
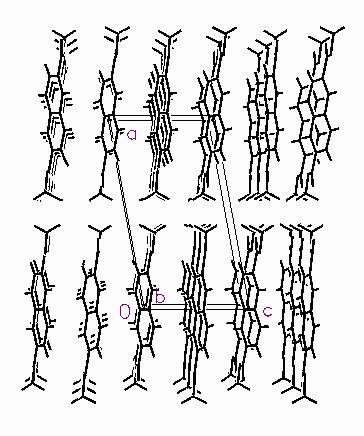 Figure 1. The crystal structure of dimethyl 2,6- naphthalenedicarboxylate, viewed down the monoclinic b-axis. Poly(ethylene 2,6-naphthalenedicarboxylate), PEN, is a promising polymer for use in applications requiring superior physical properties to those of poly(ethylene terephthalate), PET. The commercial monomer, dimethyl 2,6-naphthalenedicarboxylate (NDC) is prepared by esterification of 2,6-naphthalenedicarboxylic acid, which is prepared by homogeneous oxidation of 2,6- dimethylnaphthalene (DMN). Determining the crystal structure of our commercial material seemed like a good idea. This structure solution is an example of "trial and error". The powder pattern of NDC can be indexed on a primitive monoclinic cell, with Z = 2. The space group was unambiguously determined as P21/c. It therefore seemed likely that the molecules resided on centers of symmetry. The two shorter cell dimensions are similar to those of 2,6-dimethylnaphthalene, providing some indication of the orientation of the molecule. The NDC molecule was placed at a center, and manipulated using Cerius2 [4], simultaneously monitoring both the reasonableness of the structure (intermolecular contacts) and the agreement of the observed and calculated powder patterns. This was a humbling exercise, since it demonstrated just how sensitive the diffraction pattern is to molecular orientation! Solving the structure was complicated by the fact that there are two potential low-energy centrosymmetric conformations of the molecule, E, and Z. While a plausible (real space) structure could be obtained for the E conformation, it did not refine well. Changing to the Z conformer quickly resulted in identification of a promising low-energy structure, one which refined well. The ester groups are rotated 20° out of the plane of the naphthalene ring. This rotation "costs" about 1 Kcal/mole/ester group. The crystal structure consists of layers perpendicular to the a-axis (Figure 1). The aromatic rings are inclined, and stack parallel to c, with "side-to- side" interactions along b. These layers interact only loosely with their neighbors along a, with only end-to-end contacts between the layers. The twist of the ester groups helps the molecules interlock within the layers. The small energy penalty of the rotation results in a very efficient and beautiful packing.
Dimethyl 2,7-naphthalenedicarboxylate
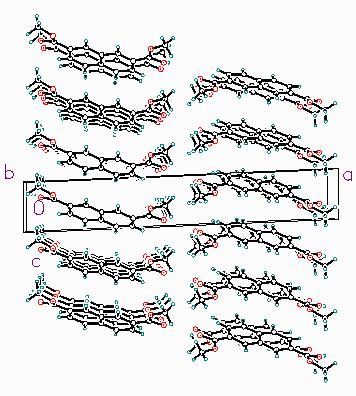 Figure 2. The crystal structure of dimethyl 2,7naphthalenedicarboxylate, viewed down the monoclinic b-axis. A common engineering practice is to modify polymer properties by incorporating a co-monomer to alter the packing of the polymer chains, and thus the physical properties of the polymer. Dimethyl 2,7- naphthalenedicarboxylate (27NDC) is of interest as a potential co-monomer in PEN applications. The crystal structure of 27NDC provides the framework for understanding the physical properties, and sheds light on how it may affect polymer chain conformations. The powder pattern could be indexed on a primitive monoclinic cell, and the space group was ambiguous. The density is such that Z = 2. Space group P21 was assumed, and verified by successful solution and refinement; no additional symmetry was detected in the refined structure. A 27NDC molecule was built in Cerius2. The ester groups were oriented in the Z conformation observed in NDC. The molecule was positioned roughly in the cell; the long a and short c cell dimensions put severe constraints on the orientation of the molecule. This structure was used as input to the STRUCTURE_SOLVE module of InsightII [5]. This module implements a Monte Carlo simulated annealing procedure, in which the metric is not energy, but the agreement of the observed and calculated power patterns. Initial runs used a rigid planar molecule; in later runs the ester torsion angles were also allowed to vary. The best solution was used as input to a Rietveld refinement. In the initial refinements, the methyl carbons were omitted; large peaks occurred in a difference Fourier map at the positions predicted by STRUCTURE_SOLVE. Both ester groups are in the Z conformation, but they are rotated only 6 and 11° out of the ring plane. The crystal structure (Figure 2) consists of layers of molecules perpendicular to a. In an individual layer, all of the 27NDC molecules are parallel, but the tilt differs in adjacent layers.
Diammonium Terephthalate
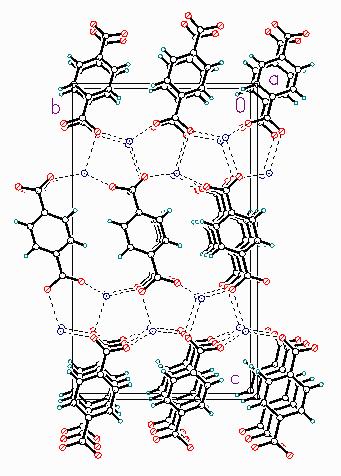 Figure 3. The crystal structure of diammonium terephthalate, viewed down the orthorhombic b- axis. Hydrogen bonds are denoted by dashed lines. The hydrogens of the ammonium ions are omitted for clarity. Amoco technology produces over 8 ´ 109 pounds of purified terephthalic acid (TA) per year. From time to time, we isolate terephthalate complexes from process streams. Understanding the natures of terephthalate complexes of catalyst and corrosion metals will lead to process insights and improvements. Nothing, however, is known about the solid-state structures of such complexes, because they are intractable solids. (The structure of a hydrated copper complex has been reported, but it’s not helpful in understanding the structures of observed process-related compounds.) We have undertaken a program to prepare and understand the solid state structures of complexes of aromatic carboxylates. Since TA is so insoluble, the typical methods of preparation involve reaction with base to prepare soluble Na or K terephthalates. These preparations involve excess base, and thus complexes are prepared at high pH. Since the oxidation of p-xylene to TA takes place in acetic acid/water, such high-pH complexes may not be relevant to process chemistry, although they are interesting in themselves. In an attempt to prepare a reagent that would permit lower-pH syntheses, I carried out a gas-solid reaction between TA and aqueous ammonia to produce diammonium terephthalate. Surprisingly, neither this compound nor its crystal structure have been reported. This structure is of interest as a model for other structures, but also because I’ve solved it three times - in three different unit cells and space groups! I’ve glossed over the determination of the unit cell in the previous structures (ITO [6] and/or DICVOL [7]), but Armel LeBail is right when he emphasizes the importance of getting the right cell! In this case, it was only the use of synchrotron data that permitted determination of the correct cell, and refinement of the structure. The synchrotron pattern can be indexed on a primitive orthorhombic cell. Laboratory patterns yielded only monoclinic or orthorhombic subcells, which permitted solution (but not refinement) of the structure. The systematic absences limited the space groups to Pbc21 or Pbcm. With Z = 4, the natural assumption is to place the TA anion at a center in Pbcm, and the one ammonium ion at a general position. The structure, however, could not be solved in Pbcm. Only when the space group was lowered to Pbc21 could the structure be solved. No additional symmetry elements are detected in the refined structure. This structure was also solved using the InsightII STRUCTURE_SOLVE module. The positions and orientations of a TA and two ammonium ions were allowed to vary/anneal, as well as the carboxyl torsion angles. After a few thousand cycles (and some false minima), a refinable model was identified. As expected, this is also a layered structure (Figure 3), with alternating hydrophobic and hydrophilic layers perpendicular to a. The C-O bonds in the carboxyl groups appear to be resonant. Each nitrogen is surrounded by four oxygens in a roughly tetrahedral arrangement; each proton of each ammonium ion participates in a hydrogen bond. It has not proved possible to refine the ammonium ions as rigid NH4 bodies; it’s amusing to watch them "tumble". In a symmetry-constrained molecular mechanics refinement with the positions of the heavy atoms fixed, the hydrogens of one ammonium ion remain pointed roughly at the surrounding oxygens, while the other ammonium ion rotates to form bifurcated hydrogen bonds. It seems that the potential energy surface is fairly flat! Within a layer, the tilts of the TA anions alternate. This structure is similar in some ways to those of potassium terephthalate and sodium terephthalate, and provides a model for the TA layers in the solid state structures of complexes of more interesting metals, as well as for TA as an intercalant in clays. We’re now applying these simulated annealing methods to the structures of transition metal terephthalates, but have encountered two factors which make this difficult. The metals so dominate the scattering that it becomes more difficult to place the terephthalates reliably. Although large numbers of metal carboxylate structure have not been reported, the observed structures exhibit a great variety of metal-carboxylate coordinations. Improvements in the force fields are clearly necessary before these tools can be used more widely. AcknowledgmentI thank Peter W. Stephens and his colleagues at beamline X3B1 at NSLS for their assistance and hospitality during the collection of the data which permitted solution of these structures.References
Please feel free to email any queries to:
r.j.cernik@dl.ac.uk
|

{kind=link}
{kind=link}