![[IUCr Home Page]](iucrhom2.gif)
![[Commission Home Page]](cpd.gif)
|
|
The IUCr-CPD Homepage is at http://www.iucr.org/iucr-top/comm/cpd/
PowderCell as teaching toolWerner Kraus and Gert NolzeFederal Institute for Materials Research and Testing (BAM), Unter den Eichen 87, D-12205 Berlin, Germany. E-mail: w.kraus@bam.de; gert.nolze@bam.de WWW: http://www.amoco.com PowderCell represents a user friendly program supporting the solution of scientific problems as well as teaching and education. Especially for the last one the program offers a lot of information given for the space-group type as well as crystal structure used. Therefore on some universities the program will be used successfully to make students familiar with X-ray crystallography (see e.g. http://wunmr.wustl.edu/Courses/Spring98/465.html) . The quasi-simultaneous diffraction pattern simulation visualizes the changes carried out on the structure. However, it is also possible to vary different diffraction parameters and investigate the resulting differences in intensity or reflection position. In principle the aim of the program is the intuitive generation of structure models.Therefore special tools have been implemented to move selected atoms or to transform the crystal structure.
The structure dataPowderCell uses structure data exported by the ICSD or Shelx. The special own data format is very simple and contains the only absolute necessary data i.e. beside the lattice parameters all atomic positions of the asymmetric unit and the space-group number corresponding to that used in the International Tables for Crystallography (HAHN (1983)). Independent from the used structure file format in the structure editor (Fig.1) volume and mass of the unit cell and the calculated X-ray density will be given. Thus also the linear absorption coefficient can be calculated by combination of these values. Of course, the coordinates of all atoms of the asymmetric unit will be listed. The used space-group number, setting and the Hermann-Mauguin symbol is shown. Nearly all data can be edited, e.g. the lattice parameters to demonstrate the reflection drifting. Atoms can be deleted or inserted to demonstrate their influence of the resulting powder diffraction pattern. Since the occupation and substitution can be varied in a simple manner, the influence on the reflection intensities can be studied. At the same time one position can be occupied and/or substituted by up to ten different elements. However, predefined ions can also be used by consideration of the specific coefficient contained in several data files.
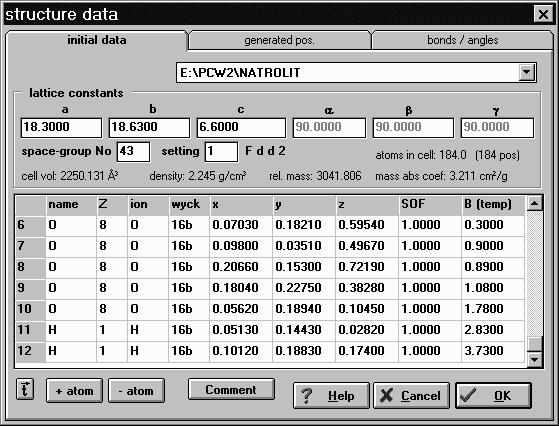 Fig.1 The structure data displays all initial data but also all generated positions and derived bonds and bonding angles. As example the structure data for Natrolite: Na2Al2Si3O10 (H2O)2 has been imported from an exported ICSD entry. Thus the influence of the different ionic states can be shown using the respective coefficients during the calculation of e.g. the atomic scattering factors. The same is valid for thermal vibrations, defined as isotropic Debye- Waller factor Biso and listed in the column B(temp). If ICSD data have been imported containing anisotropic temperature factors (Uij, bij or Bij) the data will be converted and the equivalent Debye-Waller factor Bequ will be set in the column B(temp). PowderCell is able to identify the Wyckoff positions for more than 740 different settings of the 230 space-group types. To this end approximately 210 different Wyckoff- position types have been derived following the data given in the International Tables. If one types the Wyckoff letter in the column wyck of the table shown in Fig.1 the so-called Wyckoff-position type will be given, e.g. (0,0,1/4) or (x,x,z). In the first case PowderCell inserts the coordinates immediately. In case of (x,x,z) the program combines the x and y coordinate i.e. the x value will be copied to y if one type the corresponding Wyckoff letter in column wyck. Of course, in this case the quantities must be edit manually before. The identification procedure is very comfortable if one likes to know the different special positions defined in the symmetry setting used. In future also all symmetry- equivalents of the respective special position in symbolic form shall be displayed. In the current version only the from the asymmetric unit derived symmetry-equivalent atomic positions will be given; in cartesian as well as in crystal coordinates. Sometimes the crystal structure data will be given in different settings described by the same Hermann-Mauguin symbol. In such cases the choice of the origin is defined differently. Therefore we implemented an additional translation vector t which will be combined with all atomic coordinates of the asymmetric unit (see Fig.1). Thus it is possible to input the crystal structure data and shift this using t. This enables e.g. the conversion of the two settings of several cubic and tetragonal space-group types where the origin of the used unit cell is shifted. The correspondence can be demonstrated very easy e.g. for Silicon. The crystal structure can be described by the position (1/8,1/8,1/8) using the second setting of space-group type 227. However, if one change the setting to the first and use t = ( 0.125, - 0.125, -0.125) the same structure results obvious e.g. from the identical generated powder pattern. The new version of the program offers the input of a comment (see Fig.1). If one imports ICSD files the content of this entry will be inserted automatically, but it is also possible to edit, print or cut and paste it. So small individual changes, e.g. in lattice parameters, or the structure data reference can be stored together with the data itself. Very interesting is the import of Shelx files because commonly neither the Hermann-Mauguin symbol nor the space-group number are given. Therefore PowderCell interpretes the used general positions and tries to convert this into one of the supported 740 settings. If no agreement exists the program nevertheless imports all coordinates and sets the space group on P 1; after which the user can select the correct space group from the menu. It may be possible that only an additional t must be inserted to get the accurate crystal structure description.
Unit cell transformation and subgroup generationCaused by the implementation of subgroup derivation it was necessary to consider also non-conventional settings. In principle this concerns exclusively triclinic, monoclinic, orthorhombic and rhombohedral space-group types. PowderCell enables a comfortable transformation of unit cell parameters and atomic coordinates (Fig.2).
 Fig.2 Transformation tool in PowderCell. All data will be adapted after the choice of the wished setting. This has been tested by a detailed comparisons of the resulting powder patterns. The extinction laws and the reflection generation (including the effects of multiplicity) has been included. The correlation between the different settings can be demonstrated in a simple manner using the listing of reflections considered in the chosen diffraction angle range. However, also the indexing of all symmetry- equivalent reflections can be extracted which is dependent on the respective Laue or point group. To get this one must activate the preferred orientation. Then the program creates a file orient.dat where all generated reflections will be listed among other things. Unfortunately the transformation procedure does not use the reduced cell, i.e. the calculation of the cell parameter does not follows the recommendations of the International Tables for Crystallography in each case.
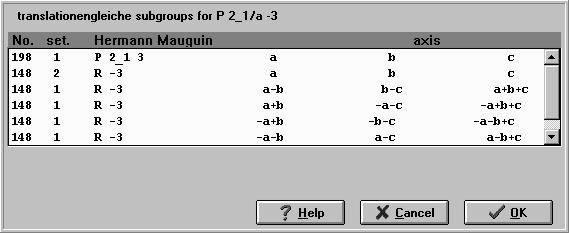 Fig.3 The subgroups have been subdivided in 4 classifications. In analogy to the transformation the existing subgroups can be chosen by selection. PowderCell represents one of the first programs which is able to derive the subgroups of a crystallographic space- group type. The used data are based on extensive derivations of MÜLLER (1994), who puts these at our disposal. The subgroup procedure allows to reduce the symmetry density of a given crystal structure. To this end all possible maximal subgroups classified in translationengleiche and klassengleiche will be displayed and the user must only select the right one (Fig.3). Also here the user of the program gets the complete transformation of the crystal structure containing the adapted lattice parameters as well as the complete number of atoms of the asymmetric unit corresponding to the new symmetry. Especially the last one is the highlight of this procedure because it simplifies the work with such problems, e.g. the investigation of phase transitions or the interpretation of the influence of lower symmetric external effects on the atomic arrangement within the crystal. Thus it should be possible to demonstrate structure similarities or to transfere one structure into another. Furthermore the Wyckoff splitting can be investigated by decreasing of symmetry density step by step. Please notice that the subgroup relations are defined for the first setting only, i.e. sometimes it is necessary to transform the derived subgroup into a conventional setting before the next decrease of symmetry can be carried out. The invariance of the resulting powder pattern is a good criterion for the correctness of the used procedure and can be observed quasi-simultaneously. The apparent change of the extinction laws, of the reflection indexing, of the multiplicities is suitable to show that a lot of crystallographic conventionts are based on the mathematical description used. The derivation of subgroups enables also the demonstration of the difference between systematic and accidental extinctions. Using the subgroup relations it is very easy to show e.g. the corellation between the different modifications of BaTiO3 observed in former days and described by the space groups P m3m, P 4mm, R 3m and A mm2. Thus it is really possible to derive the different modifications from the Perowskite structure (P m3m). However, also ordered strcutures like Ni3Al (g‘ precipitates in nickel-base superalloys - structure type: L12) can be extracted from the disordered structure of the g phase: Ni0.75Al0.25 (structure type: A1).
Structure manipulationThe lost of symmetry caused by derivation of subgroups is equivalent to a decrease of local symmetry, i.e. an atom located on a special position will be transfered to a location described by a more generally position. The result is that often the atom is not more fixed on this position but gets the ability to move within the unit cell. This has been carried out in PowderCell. To this end parts of the crystal structure must be selected. These will be visualized as hatched atoms. The tools for moving allow to rotate or translate the selected parts. Thus it is possible to shift along the basis vectors or along a direction defined by two atoms. Rotations are allowed around a given atom, the center of gravity of selected atoms or arround a direction defined by two atoms again. Until now the manipulation is not Wyckoff position sensitive i.e. it is impossible to translate or rotate under condition of preservation of the local symmetry. Quite the reserve, the symmetry of the new position will be analysed again and all symmetry-equivalent positions will be generated. However, especially for low-symmetry crystal structures the given manipulation tools enables a successful variation of structure models what has been proved e.g. by RECK (1994). For education it is useful to demonstrate the influence of local symmetry on the crystal structure but also on the resulting powder pattern. So one can study the influence of the the position of heavy atoms within the unit cell or the dependence of the orientation of molecule fragments etc. (see http://www.bam.de/a_v/v_1/powder/e_cell.html).
Powder diffraction simulationThe different examples given below show clearly that the advantage of PowderCell is based on the combination of structure visualizer and powder pattern generation. Even of the last one we maked a point because from the comparison of experimental and theoretical curve results the quality criterion for the used or created structure model. In contrast to that the structure viewer really represents only the means to an end. The generation of the powder pattern considers several parameters which influence the resulting diffractogram - Fig.4. The most important fact is that these parameters can be changed very user friendly. On the work sheet experiment the X-radiation can be selected from a set of 11 different anode materials, the anomaleous dispersion or the a1/a2 doublet can be switched on/off, the angle range can be set as wished. Thus the influenceof the different parameter can be studied intensively, especially if one considers additionally that these are dependent on the used crystal symmetry, lattice parameters and element distribution. However, the resulting reflection intensities must be convoluted using suitable profile functions. Furthermore the change of the peak width in dependence on the diffraction angle must be considered by the use of a 3 parametric model function (CAGLIOTI (1958)). On the other hand preferred orientations influence the integral intensity thereby that the effective multiplicity varies.
 Fig. 4 Free definable parameters for the powder diffraction simulation in dependence on each phase. The phase- independent, general parameters can be set in experiment. From these a lot of demonstrations are imaginable e.g. how does the use of variable slit system or primary monochromator influences the powder pattern; when the anomaleous dispersion must be considered; how one may detect the character of preferred orientation, how differs X- ray and neutron diffraction patterns etc. Very often these are very practice-oriented questions, so that the simulations of such problems may support the teaching very effectively.
Data and graphical outputPowderCell enables the user to print directly all data displayed or listed in the program in any form, e.g. the structure data, the comment, the reflection table and the used experimental conditions. Furthermore it is possible to print the crystal structure as well as the powder pattern directly. However, it is more important to export the graphs because in PowderCell one cannot edit the pictures (Fig. 5). For the crystal structure representation the use of POVRay, a freeware raytracing program, has been supported (www.povray.org). POVRay generates photorealistic pictures using the exported scripts (Fig. 5).
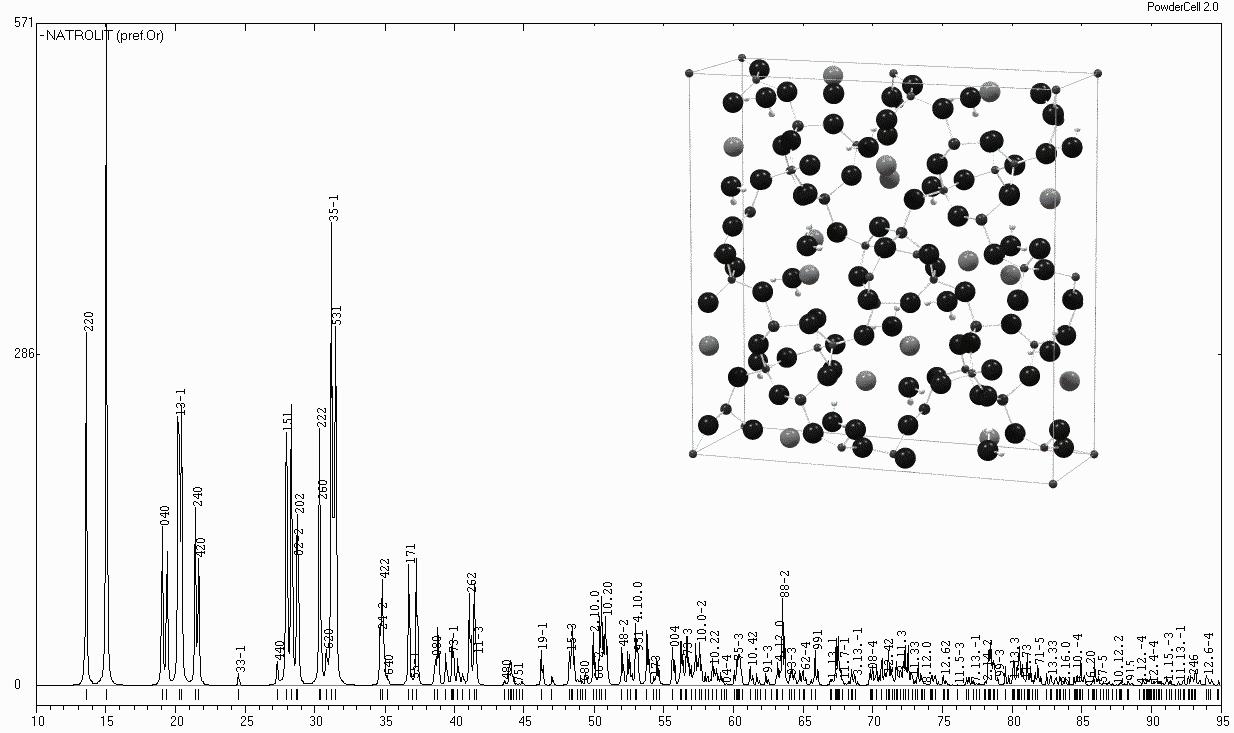 Fig. 5 Calculated X-ray powder pattern of Natrolithe exported by PowderCell. The displayed structure picture has been created using POVRay.
System requirementsThe program runs under Microsoft Windows 3.1, 95 and NT. Because of the complex calculations a Pentium CPU will be recommended. PowderCell needs approximately 5 Mbyte on harddisk and more than 4Mbyte main memory. The program is free of charge and can be downloaded from http://www.bam.de/a_v/v_1/powder/e_cell.html References
Please feel free to email any queries to:
r.j.cernik@dl.ac.uk
|

{kind=link}
{kind=link}