![[IUCr Home Page]](iucrhom2.gif)
![[Commission Home Page]](cpd.gif)
|
|
The IUCr-CPD Homepage is at http://www.iucr.org/iucr-top/comm/cpd/
BGMN - a New Fundamental Parameters Based Rietveld Program for Laboratory X-ray Sources, it's Use in Quantitative Analysis and Structure InvestigationsNorman Binsted, Maria Pack, Mark Weller, and John EvansDepartment of Chemistry, University of Southampton, Southampton SO17 1BJ, UK. E-mail: N.Binsted@dl.ac.uk; mtw@soton.ac.uk; je@soton.ac.uk; WWW: http://www.soton.ac.uk/~chemweb/
The complimentary nature of the techniques of EXAFS and X-ray or neutron powder diffraction (PD) often leads to their combined usage in the study of crystalline materials. For example, X-ray PD is very good at determining the coordinates of heavy atoms in special positions, but less good at their oxygen neighbours. Metal-edge EXAFS may then provide useful constraints on M-O distances. There are instances, for example in mixed-metal periodates such as RbGeIO6 [1], where the reflections due to the metal sites exactly overlap (1/3,2/3,1/2 and 2/3,1/3,1/2 sites in P312). The sites differ in their oxygen coordination distance however, so EXAFS can uniquely resolve them. In other instances the short range order seen by EXAFS contrasts with the long range order seen by PD. Rutile (TiO2) may accommodate high concentrations of elements such as Ta or Nb, without any change in overall lattice symmetry but EXAFS would reveal different bonding distances for Ti, Nb and Ta due to local distortion of the oxygen framework. In these cases, EXAFS may provide significant additional information, but is not being used directly to improve the refined positional coordinates. Moreover, non-nearest neighbour information is not fully utilised. EXAFS analysis usually involves refinement of a series of shell distances, perhaps including a few multiple scattering (MS) paths for in-line atoms. In practice, non-nearest neighbour peaks almost never correspond to simple shells of atoms. Structure to 5 or 6 A may contain dozens of different distances and thousands of MS paths, which are not well represented by a small set of Gaussian shells. It is also very unlikely that a refined set of spherical polar coordinates is compatible with any possible crystal structure. For these reasons, a program was developed at Southampton [2] which uses a single model of the crystal structure to fit both PD and EXAFS spectra (it has been used for up to five different edges from Gd2Ba2CaCu2Ti3O14 simultaneously [3]). By modifying the treatment of disorder used in EXCURV92 (and most other codes) systematic differences between the techniques have been minimised, so that in most cases the two methods 'agree' on the correct solution. The PD profile essentially determines the symmetry and cell parameters, but the oxygen positions and distribution of metal atoms over sites is strongly dependent on the EXAFS. Refinement of the structure requires only a few more parameters than a Rietveld refinement - one edge offset per EXAFS spectrum, plus either some EXAFS Debye-Waller factors or a small set of adjustable parameters from which they may be calculated (in [3], two parameters were used to approximate the DW terms for over three hundred shells and all the MS paths between them). The EXAFS spectra may then be fitted as far out in R-space as time permits (106 path MS calculations are still slow).
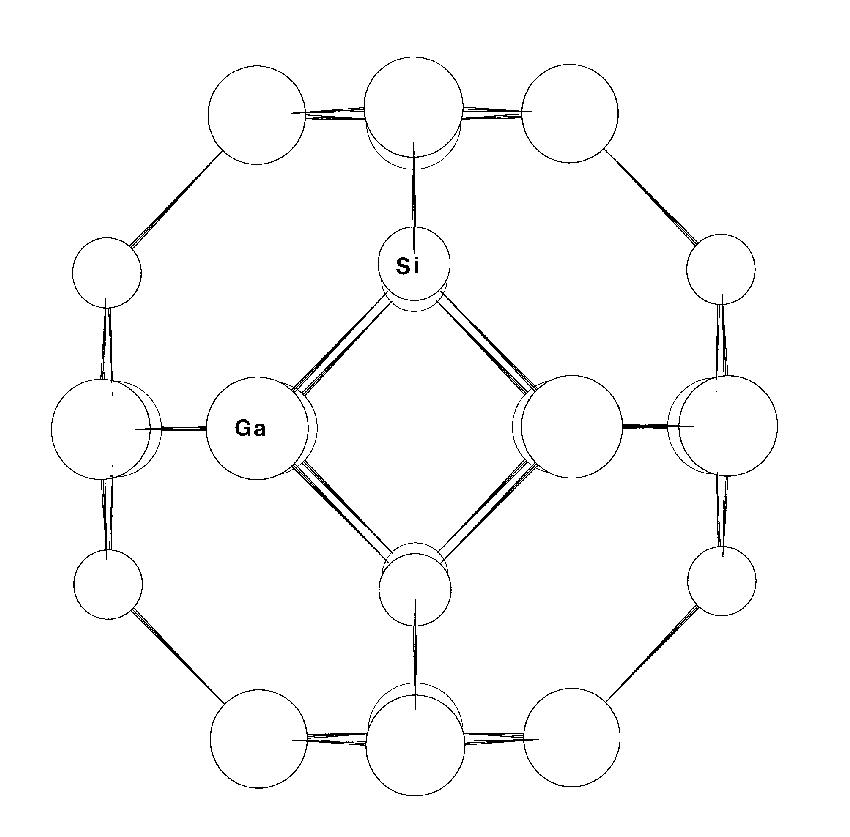 Figure 1: The framework structure of Gallobicchulite Ca8(Ga2SiO6)4(OH)8 (oxygens excluded). One of the most interesting applications of the method was to Gallobicchulite [4], Ca8(Ga2SiO6)4(OH)8, a synthetic sodalite. The structure is that of a *-cage zeolite, with four-membered rings of tetrahedrally coordinated cations (T), linked to form six-membered rings normal to the (111) directions of the cubic cell, and with intrachannel Ca and OH on cube diagonals (fig.1). In this material NMR and EXAFS data [5] suggested a high degree of ordering in terms of (Si,Ga) occupancy of sites and Ga-O bond lengths consistent with distinct gallium sites. The powder diffraction data, however, indicated a structure disordered on a large scale, with a single T-O distance.
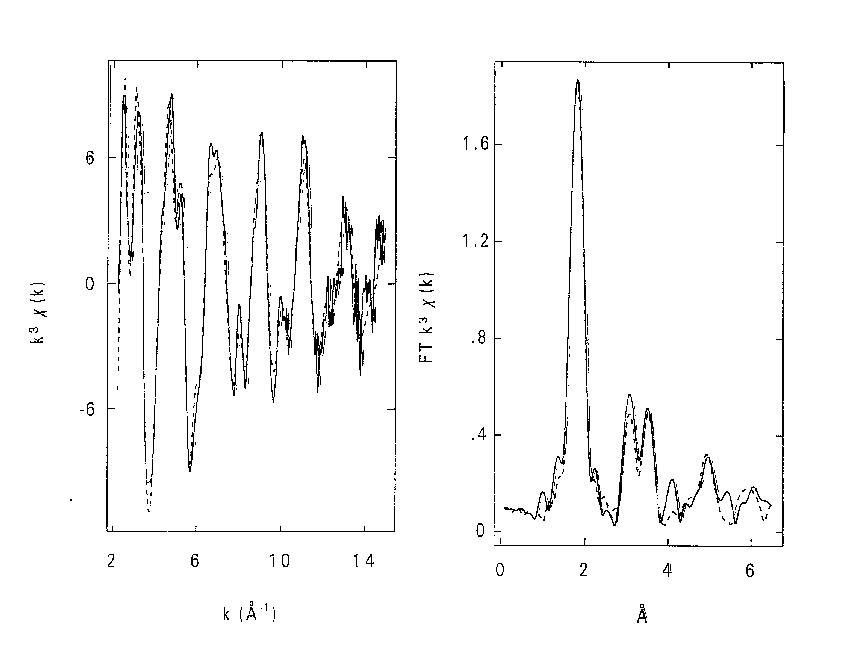
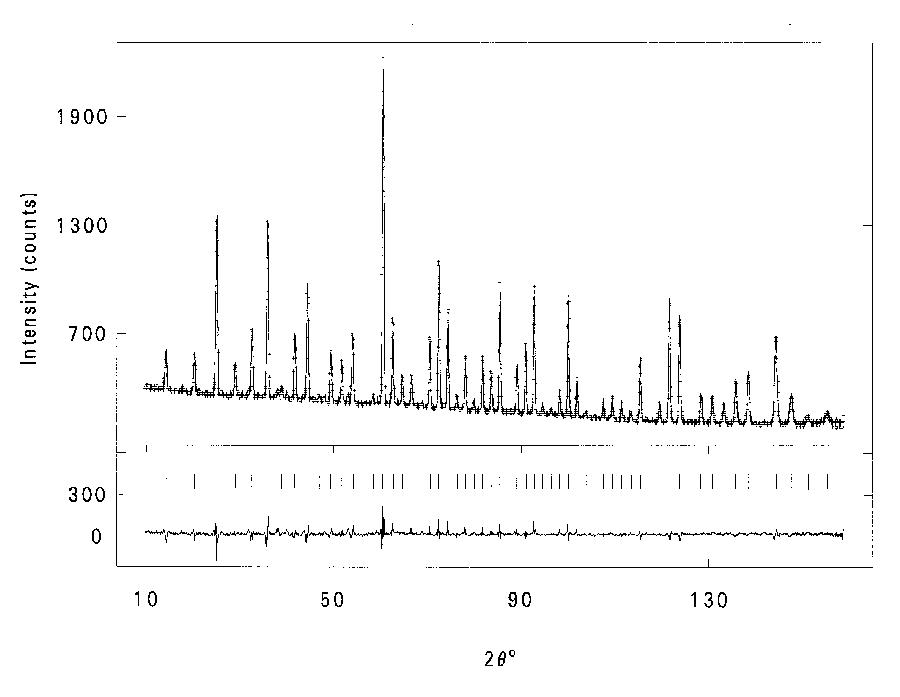 Figure 2: Fits to Ca8(Ga2SiO6)4(OH)8 using combined refinement with space group I-4 for EXAFS and I-43m for neutron data. a). Fit to k3 weighted Ga K-edge EXAFS data (left) and Fourier transform (right), phase- corrected for Ga and O. b). Fit to powder neutron diffraction profile. The combined program gave a fit to both neutron PD and EXAFS with an identical set of refined coordinates however (fig.2), although this was only achieved by using two different space groups. With I-4 the coordinates produce a pseudo-cubic structure with framework ordering similar to the sodalite-mineral tugtupite [6]. This fits the EXAFS, explains the NMR data, and is consistent with Löwensteins rule [7]. With I-43m (and fractional occupancies of some sites) it generates the PD profile. The relationship between the two cells is exactly equivalent to averaging over x, y and z orientations of the unique axis in I-4. We interpret the structure as consisting of domains with one of three possible orientations within the crystal. The method has thus yielded a set of positional parameters for a basic cell that is 'invisible' to diffraction methods. It is hoped to further develop the program in several ways. One is to extend the modelling of local order - for example by using supercells with lattice distortions for mixed-metal oxide EXAFS while using the average over constituent cells for PD. Another is to achieve a common treatment of disorder. The disorder parameters should agree between EXAFS and PD as given by:
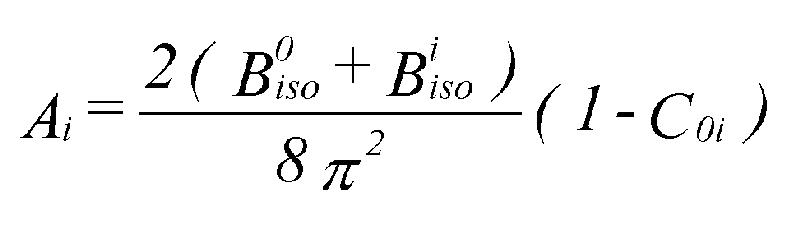 A is the EXAFS Debye-Waller factor for shell i, and C0i the correlation between shell i and the central atom. Provided the correlations can be obtained and systematic errors in PD thermal parameters can be eliminated (systematic errors do not seem to be a problem with EXAFS), a common set of refined thermal parameters can be extracted. This would help reduce correlation between positions and Debye-Waller factors, so giving more reliable structures, and also help to distinguish between short range and long range disorder. Acknowledgements. We are grateful to the EPSRC for funding this work, to Daresbury Laboratory for allowing the use of code from EXCURVE, and to DL theory group for help with computing equipment. The program uses code from DBW [8], FEFF [9], and the UKAEA Harwell library [10].
|

{kind=link}
{kind=link}